

Научный руководитель Института катализа СО РАН академик Валентин Николаевич Пармон стал лауреатом премии «Глобальная энергия». Премия была присуждена за прорывную разработку новых катализаторов в области нефтепереработки и возобновляемых источников энергии, внесших вклад в развитие энергетики будущего.
Премия «Глобальная энергия» учреждена в 2003 году. Она присуждается за выдающиеся исследования и научно-технические разработки в области энергетики, которые способствуют эффективному использованию энергетических ресурсов и экологической безопасности на Земле в интересах всего человечества. С момента учреждения ее лауреатами стали 33 ученых из Великобритании, Германии, Исландии, Канады, России, США, Франции, Украины, Швеции и Японии.
Решение по выбору лауреатов премии принимает Международный комитет по присуждению премии «Глобальная энергия», в состав которого входят 20 авторитетных ученых из 13 стран. Номинационный пул премии – более 3000 ученых из 83 стран.
Премия учреждена в России при поддержке ведущих российских энергетических компаний – Газпром, Сургутнефтегаз и ФСК ЕЭС.
Указом Президента Франции Франсуа Олланда академик В.Н. Пармон удостоен высокой государственной награды Французской Республики – почетного звания Кавалера Национального Ордена «За заслуги». Об этом в своем письме новому Кавалеру Ордена сообщил Посол Франции в Российской Федерации, господин Жан-Морис Рипер. Посол высказал свои поздравления и отметил «неоценимый вклад» Валентина Николаевича Пармона «в упрочение научного сотрудничества и обменов между нашими странами».
Орден «За заслуги» (Ordre national du Mérite) – национальный орден Франции, учреждённый в 1963 году Президентом Франции Шарлем де Голлем в результате реформирования системы высших наград страны, является вторым по значимости универсальным национальным орденом после Ордена Почетного Легиона. Он вручается за выдающиеся заслуги перед Францией, как французским гражданам, так и иностранцам, людям различных профессий, гражданским и военным.
Научный совет по катализу ОХНМ РАН поздравляет Валентина Николаевича с высокими наградами и желает новых творческих достижений!

28 июня исполнилось 85 лет действительному члену Российской академии наук, Советнику РАН Владимиру Борисовичу Казанскому.
В.Б. Казанский окончил Химический факультет МГУ в 1954 году. С 1955 по 1966 год работал младшим и старшим научным сотрудником Института химической физики АН СССР.
С 1966 г. — зам. директора, зав. лабораторией спектральных и квантово-механических исследований каталитических реакций, зав. лабораторией радиоспектроскопических и оптических методов изучения механизма гетерогенного катализа Института органической химии им. Н.Д. Зелинского РАН.
Член-корреспондент c 1974 г., академик c 1991 г. — Отделение химии и наук о материалах.
В.Б. Казанский — крупный ученый в области катализа, спектроскопии, квантовой химии, химии и физики поверхности.
Область научных интересов: физическая химия, изучение механизма гетерогенного катализа, спектральные исследования в области структуры активных центров катализаторов и промежуточных состояний реакционных частиц.
Разработал основы катализа (установление взаимосвязи между строением активных центров гомогенных и гетерогенных катализаторов, природой образующихся с их участием активных интермедиатов и детальным механизмом каталитических реакций). В 1960-е годы им были выполнены работы по изучению механизма гидрирования олефинов на металлах с использованием дейтерия.
Ввел в практику исследований гетерогенных катализаторов парамагнитный резонанс, ультрафиолетовую и ИК-спектроскопию диффузионного отражения, ядерный магнитный резонанс высокого разрешения.
Экспериментально изучил свойства поверхностных низкокоординированных ионов переходных металлов и установил их роль в катализе на оксидах. Исследовал механизм образования на поверхности катализаторов свободных радикалов при адсорбции и фотодиссоциации различных молекул. Определил природу избыточной каталитической активности, возникающей при ионизирующем облучении оксидных катализаторов, и выяснил, какую роль играют в радиационном катализе кислородные «дырочные» центры. Разработал новые спектральные методы изучения бренстедовской кислотности гетерогенных катализаторов. Выполнил квантово-химические расчеты структуры активных центров и состояния адсорбированных молекул.
Более поздние работы посвящены исследованию роли сольватации в кислотно-каталитических реакциях.
В.Б. Казанский создал свою научную школу — среди его учеников более 40 докторов и кандидатов наук.
Автор около 700 научных работ, обзоров и патентов.
Главный редактор журнала «Кинетика и катализ».
Член бюро Научного совета РАН по катализу.
Награжден орденом «Знак Почета».
Лауреат премии им. А. Гумбольдта.
Научный совет по катализу ОХНМ РАН и редакция Каталитического бюллетеня
сердечно поздравляют
Владимира Борисовича с юбилеем и желают крепкого
здоровья, успехов и благополучия!

15 мая исполнилось 70 лет члену-корреспонденту РАН Усеину Меметовичу Джемилеву.
У.М. Джемилев с отличием закончил Казахский химико-технологический институт. В 1969 году поступил в аспирантуру Института химии Башкирского филиала АН СССР, которую закончил досрочно. В 1972 году защитил кандидатскую диссертацию, в 1977 году – диссертацию на соискание ученой степени доктора химических наук на тему “Исследование в области синтеза непредельных соединений с участием комплексов переходных металлов” на специализированном Совете по защите докторских диссертаций при ИОХ им. Н.Д. Зелинского АН СССР. В 1977-1992 гг. работал заведующим лабораторией Института химии БФ АН СССР (с 1987 г. ИХ БНЦ УрО АН СССР). В 1981-1992 гг. – заместитель директора по научной работе Института химии БФ АН СССР. В 1983 году ему было присвоено ученое звание Профессор по специальности “Органическая химия”. В 1990 году избран членом-корреспондентом АН СССР по Отделению общей и технической химии АН СССР. С 1993 – заместитель Председателя Президиума Уфимского научного центра РАН, с 2011 г. – Председатель УНЦ РАН.
С 1992 г. – директор Института нефтехимии и катализа РАН (г. Уфа). Лауреат Государственной премии СССР (1990 г.), Лауреат Государственной премии РФ в области науки и техники за 2003 год (2004 г.), награжден медалью “Ордена за заслуги перед Отечеством II степени” (1999 г.). Руководитель научной школы по металлокомплексному катализу, член редколлегий научных журналов РАН "Органическая химия" и "Нефтехимия", член Научных советов по катализу РАН и нефтехимии РАН, автор и соавтор свыше 600 научных статей и обзоров, 656 авторских свидетельств и патентов, 2 монографий, 2 книг-обзоров, изданных за рубежом, научный руководитель 10 докторских и 65 кандидатских диссертаций.
Мировую известность ему принесли работы в области комплексного циркониевого катализа органических и металлоорганических реакций.
У.М. Джемилев внес крупный вклад в становление и развитие фундаментальных исследований в области каталитической активизации атомов, малых и малостабильных молекул, теломеризации сопряженных диенов, синтеза гетероциклов и гетероатомных соединений уникальной структуры, линейной и циклической олигомеризации диенов и олефинов, в том числе асимметрической.
Им разработаны каталитические методы синтеза ранее неизвестных классов металлоорганических соединений, что положило начало развитию новой области химии — химии металлоциклов непереходных металлов.
Совместно с лабораторией академика О.М. Нефедова разработана новая стратегия одностадийного каталитического синтеза высоконапряженных полициклических структур, открыты новые общие реакции конструирования соединений, построенных из трех-, четырех- и пятичленных циклов, изучена химия этого класса соединений.
Разработаны фундаментальные реакции, новые общие принципы и методы гетероциклизации сопряженных диенов и ацетиленов с получением широкого класса практически важных кислород-, азот- и серосодержащих гетероциклов.
Открытые У.М. Джемилевым реакции этилмагнирования и циклоалюминирования непредельных соединений используются в мировой практике как именные реакции («Реакции Джемилева»).
При личном участии У.М. Джемилева разработаны и внедрены в промышленность технологии получения новых мономеров, нейтрализаторов сероводорода, ингибиторов коррозии, смазочно-охлаждающих жидкостей, современных препаратов для медицины и сельского хозяйства, продуктов и материалов с рекордными характеристиками для специальной техники.
Научный совет по катализу ОХНМ РАН и редакция Каталитического бюллетеня сердечно поздравляют Усеина Меметовича с юбилеем, желают ему дальнейших успехов на благо российской науки!
17-23 апреля 2016 г., Белград, Сербия

17-23 апреля 2016 года в конференц-центре отеля ZIRA (Белград, Сербия) состоялся II Научно-технологический симпозиум «Нефтепереработка: катализаторы и гидропроцессы». Основными организаторами симпозиума выступили Институт катализа им. Г.К. Борескова СО РАН, Институт проблем переработки углеводородов СО РАН и ПАО «Газпром нефть».
В настоящее время наблюдается стремительный рост исследований в области каталитических гидрогенизационных технологий нефтепереработки. Наиболее важной составляющей развития данного направления являются разработка и применение новых катализаторов для осуществления гидропроцессов. Первый симпозиум по этой тематике, прошедший в Санкт-Петербурге в 2014 году, продемонстрировал интерес к достижениям в сфере разработки новых катализаторов и процессов для нефтепереработки, а также тенденциям технологического развития этих отраслей в России и мире. Было принято решение расширить круг экспертов, заинтересованных в обсуждении современных тенденций в развитии каталитических гидрогенизационных технологий, организовывая симпозиум в других странах и придав ему статус международного.
Основной задачей прошедшего симпозиума стало обсуждение представлений о создании перспективных катализаторов для процессов нефтепереработки; обмен мнениями между научными специалистами и представителями промышленных компаний о достижениях в разработке новейших катализаторов и их широком использовании; анализ тенденций развития отрасли и выработка единых подходов по внедрению в промышленность конкурентоспособных инновационных технологий.
II Научно-технологический симпозиум «Нефтепереработка: катализаторы и гидропроцессы» собрал 80 участников из 10 стран. Наряду с представителями России, в симпозиуме также приняли участие специалисты из Великобритании, Германии, Греции, Катара, Мексики, Польши, Сербии, США и Франции.

На симпозиуме было представлено 7 пленарных и 5 ключевых лекций, 30 устных секционных докладов, 16 стендовых докладов по следующим направлениям:
Разработка и совершенствование катализаторов для осуществления гидропроцессов возможны только при наличии представлений о строении активных компонентов катализаторов и механизмах их действия, которые формируются на основании фундаментальных исследований. Поэтому большая часть докладов симпозиума была посвящена фундаментальным аспектам синтеза, характеризации и исследования катализаторов процессов гидроочистки, гидроизомеризации, гидрокрекинга и пр.

При разработке и оптимизации гидрогенизационных процессов весьма важны также теоретические вопросы инженерно-технологического характера, в особенности исследование кинетики гидрогенизационных реакций и математическое моделирование реакторов для их осуществления, исследование фундаментальных аспектов тепло- и массообмена в слоях катализаторов.
Таким образом, особенность тематики симпозиума заключалась в том, что при очевидной практической направленности многие сообщения, включая пленарные и ключевые лекции, были посвящены фундаментальным основам гидропроцессов.
Участниками симпозиума был отмечен высокий уровень пленарной сессии. Первым был представлен доклад профессора Милорада Дудуковича (Университет Вашингтона в Сент Луисе, США) «Мультимасштабный инжиниринг в гидропереработке». В ходе лекции было справедливо отмечено, что переработка нефти остается основным источником транспортного топлива во всем мире. На протяжении многих лет отрасль адаптировалась к изменяющимся источникам исходного сырья. Были изобретены и реализованы многочисленные крупномасштабные каталитические процессы в производстве различных видов топлива для удовлетворения потребностей рынка. Успех переработки нефти в создании желаемых продуктов основан на серии гидропроцессов, для большинства которых требуются твердые катализаторы и надежные методы для выделения водорода.

Современные заводы, которые часто называют «соборами химической инженерии», являются прекрасными примерами торжества систематического применения методологии проектирования химических реакций (CRE) различных уровней сложности. Поэтому переработка нефти обладает одним из лучших технологических факторов Е (т.е. низким показателем нежелательных побочных продуктов на единицу желаемого произведенного продукта). В лекции проанализированы предпосылки успешного развития гидропроцессов и намечены перспективы разработки новых катализаторов и реакторов. Была продемонстрирована необходимость систематического применения многоуровневого анализа взаимодействия реакционных превращений и транспорта реагентов в различных масштабах: от микроустановок до крупных заводов. Сейчас достижения в области разработки катализаторов часто ограничиваются использованием их в существующих типах реакторов, где их потенциал не может быть полностью реализован. Также было показано, как передовые экспериментальные технологии изучения структуры потоков могут быть использованы для усовершенствования реакторов с подвижным и псевдоожиженным слоем катализатора.

Профессор Дудукович суммировал данные о том, какие разработки необходимы, чтобы сформировать улучшенную научную базу для операций гидроочистки, которые будут способствовать увеличению производительности процессов. Он отметил также необходимость подготовки новых поколений для работы в этой важной области, в частности, путем вовлечения молодых ученых в конкурентоспособные исследования с участием представителей научно-исследовательских институтов и промышленности.
Продолжил пленарную сессию профессор Валентин Николаевич Пармон (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск, Россия) с лекцией «Перспективные направления и тенденции в нефтепереработке: катализаторы и технологии».

Было показано, что каталитические технологии являются ключом к глубокой переработке углеводородного сырья. Индекс сложности нефтепереработки (так называемый индекс Нельсона) демонстрирует превосходство того или иного завода, занимающегося переработкой сырой нефти. Низкий индекс Нельсона характеризует простейшие физические и тепловые процессы в нефтепереработке и нефтехимии, в то время как каталитические процессы могут обеспечить глубокую переработку и, соответственно, высокий коэффициент Нельсона. Ключевым элементом в продвижении каталитических процессов является разработка новых катализаторов для нефтепереработки и нефтехимии.

Представленные профессором Пармоном данные относятся к развитию новых катализаторов для ряда процессов гидрогенизационной переработки нефтяных фракций, а также новых катализаторов и процессов нефтехимии. В частности, были описаны катализаторы селективной гидроочистки бензина каталитического крекинга (БКК), которые позволяют проводить гидроочистку всего БКК, без его предварительного разделения на лёгкую и тяжёлую фракции, и обеспечивают достижение остаточного содержания серы не более 10 м.д. при снижении исследовательского октанового числа не более чем на 1 пункт; катализатор для гидроочистки прямогонных и смесевых дизельных фракций до 10 м.д. серы при температуре на входе в реактор не более 340°C; катализаторы гидроочистки вакуумного газойля с получением сырья каталитического крекинга с остаточным содержанием серы менее 200 м.д. с повышенным на 5-10% выходом целевой фракции при снижении температуры процесса на 5-10°C катализаторы гидрокрекинга вакуумного газойля (ВГО), обеспечивающие выход средних дистиллятов (керосин и дизельное топливо) на 3-5% выше, чем на известных катализаторах.
Разработаны катализаторы для вакуумного дегидрирования легких углеводородов, катализаторы переработки этанола в нефтехимические продукты – для селективного окисления этанола до уксусной кислоты и каталитической переработки этанола до сверхвысокомолекулярного полиэтилена (СВМПЭ) и многослойных углеродных нанотрубок (МУНТ). В качестве примеров были приведены композитные материалы на основе СВМПЭ и МУНТ, а также некоторые данные по промышленному применению новых катализаторов.
Представитель Honeywell UOP (Дес-Плейнс, Иллинойс, США) доктор Курт ВанденБуш выступил с лекцией «Гидропереработка: набор разноплановых технологий для удовлетворения быстро эволюционирующих потребностей производителей топлива». В выступлении было отмечено, что волатильность рынка ископаемого сырья и ужесточение технических характеристик предельно допустимых параметров выброса в атмосферу SOx, CO2 и сажи, связанных с потреблением транспортного топлива, приводят к постановке массы новых интересных задач перед учеными в области разработки комплекса технологий гидропроцессов. Разработка сланцевого сырья также приводит к диверсификации производств с постановкой новых задач по обработке тяжелых парафинов, что может обеспечить вовлечение в переработку новых типов сырья, а также позволит перерабатывать более тяжелые нефтяные фракции.

В своей презентации доктор ВанденБуш наметил ряд макроэкономических тенденций и продемонстрировал их непосредственное влияние на развитие технологии гидропроцессов. Был сделан обзор самых передовых технологий гидропроцессов, а затем представлены некоторые ключевые достижения в области фундаментального понимания функциональных возможностей катализа, характеризации исходного сырья, а также метода моделирования процессов в указанной области. По мнению доктора ВанденБуша, этот научный прогресс, расширенные возможности моделирования и применение подхода, основанного на изучении процессов на молекулярном уровне, вдохновляли и продолжают вдохновлять ученых на усовершенствование технологий нефтепереработки. В лекции были приведены примеры успешного технологического развития в трех тематических областях: конверсия среднего дистиллята из ископаемых источников, последние достижения в области конверсии тяжелой нефти в топливо и коммерциализация производства топлив из смешанных биоисточников.
Продолжила пленарную сессию профессор Ирина Игоревна Иванова (Московский государственный университет им. М.В. Ломоносова, Москва, Россия) с презентацией «Разработка микро-мезопористых катализаторов на основе цеолитов для нефтехимии и нефтепереработки».

Ее лекция была посвящена микро-мезопористым материалам, новым процессам рекристаллизации цеолитов, которые были продемонстрированы в качестве универсальных инструментов для специализированного синтеза мезопористых цеолитов, микро-мезопористых нанокомпозитов и упорядоченных мезопористых материалов с цеолитными фрагментами в стенках.
Иерархические микро-мезопористые материалы появились в качестве важного класса каталитических материалов, которые имеют значительные преимущества по сравнению как с классическими цеолитами, так и с мезопористыми нецеолитными материалами. В настоящее время этот класс материалов включает различные структурные типы: наноразмерные цеолиты с мезопорами между кристаллитами, расслоившиеся цеолиты, цеолитные нанолисты, мезопористые цеолиты, упорядоченные мезопористые материалы с полностью или частично кристаллическими цеолитными стенками, микро-мезопористые композитные материалы и т.д. Рекристаллизованные материалы подразделяются на три отчетливо различимые группы в зависимости от степени рекристаллизации: I) мезопористые цеолиты (RZEO-1), II) микро-мезопористые нанокомпозиты (RZEO-2), III) мезопористые материалы с цеолитными фрагментами в стенках (RZEO-3).
Первая часть лекции была сосредоточена на механизме рекристаллизации цеолитов, которая определяет основные синтетические стратегии, ведущие к получению различных типов рекристаллизованных материалов. Во второй части было рассмотрено влияние структуры, текстуры и пористости рекристаллизованных цеолитов на их кислотные и диффузионные свойства. Третья часть была посвящена технике рекристаллизации материалов для конкретных каталитических систем применительно к нефтехимии и нефтепереработке. Особое внимание было уделено влиянию рекристаллизации на улучшение каталитической активности цеолитов и мезопористых материалов в различных каталитических реакциях. В последней части лекции были рассмотрены преимущества и недостатки микро-мезопористых материалов, полученных при рекристаллизации цеолитов, относительно чистых микро- и мезопористых молекулярных решеток и других иерархических цеолитов, а также проанализированы возможности и перспективы развития данной технологии.
Профессор Драгомир Букур (Texas A&M University at Qatar, Доха, Катар) представил доклад «Переработка синтез-газа, полученного из природного газа, угля и биомассы, в транспортное топливо и химическое сырье через синтез Фишера-Тропша».

Возобновление интереса к конверсии синтез-газа в углеводороды с использованием синтеза Фишера-Тропша (ФТ) вызвано главным образом опасениями скорого исчерпания запасов традиционных ископаемых видов топлива и необходимостью использования возобновляемых источников сырья. Сырье, полученное как из ископаемых, так и из возобновляемых ресурсов, может быть переработано в жидкое топливо и химическое сырье с использованием технологий ХВЖ (X-В-Жидкость) технологии, где X может быть природным, попутным или сланцевым газом, углем или биомассой. Основой процессов газ-в-жидкость, биогаз-в-жидкость и уголь-в-жидкость является реакция синтеза Фишера-Тропша, в ходе которой синтез-газ превращается в углеводороды, с применением кобальтовых или железосодержащих катализаторов. Кобальтовый катализатор применяется при низких температурах и высоком соотношении Н2/СО в подаваемом сырье. Катализаторы на основе железа используются для переработки угля, получения синтез-газа с низким соотношением H2/CO.
Существуют два основных типа технологии ФТ, которые в настоящее время применяются в промышленных масштабах: высоко- и низкотемпературный синтез Фишера-Тропша, где последний, как правило, используется в промышленности для синтеза жидкого топлива. Низкотемпературный синтез Фишера-Тропша является трехфазной операцией и в основном проводится в двух типах коммерческих реакторов: реакторах со взвешенным слоем мелкодисперсного катализатора и многотрубчатых реакторах с неподвижным слоем гранулированного катализатора. Промышленные реакторы для высокотемпературного синтеза Фишера-Тропша должны выбираться с учетом того, что данная реакция сильно экзотермическая и необходимы системы отвода тепла. Другие вопросы, которые также должны учитываться, это эффективность катализатора, дезактивация и регенерация катализатора, снижение давления и т.д. Ожидается, что небольшие блоки переработки биогаза в жидкие топлива могут оказаться эффективными с экономической точки зрения. Кроме этого, в лекции были рассмотрены последние события и тенденции в области технологии ХВЖ, в частности, дан краткий обзор кинетических подходов к моделированию для основных и побочных реакций.
В третий рабочий день симпозиума были представлены две заключительные пленарные лекции, одна из которых была сделана профессором Хорхе Анчета из Мексиканского Института нефти (Мехико, Мексика): «Каталитические гидропроцессы для переработки тяжелых нефтей и остатков».
 В своей лекции профессор Анчета презентовал метод HIDRO-IMP®. Это каталитическая технология гидроочистки для переработки тяжелых и сверхтяжелых нефтей с целью улучшения их текучести и снижения вязкости для обеспечения лучшего качества сырья для нефтеперерабатывающих заводов. Производимая модернизированным методом синтетическая нефть близка по своим свойствам к традиционной сырой нефти, но содержит меньше серы и других примесей. Главной особенностью процесса HIDRO-IMP® является использование серии последовательно расположенных реакторов с неподвижным слоем катализатора со специально подобранной системой загрузки слоев, состоящих из селективных катализаторов для гидроочистки и гидроконверсии в сочетании с умеренным давлением процесса, что сводит к минимуму образование остаточных продуктов и углеродистых отложений.
В своей лекции профессор Анчета презентовал метод HIDRO-IMP®. Это каталитическая технология гидроочистки для переработки тяжелых и сверхтяжелых нефтей с целью улучшения их текучести и снижения вязкости для обеспечения лучшего качества сырья для нефтеперерабатывающих заводов. Производимая модернизированным методом синтетическая нефть близка по своим свойствам к традиционной сырой нефти, но содержит меньше серы и других примесей. Главной особенностью процесса HIDRO-IMP® является использование серии последовательно расположенных реакторов с неподвижным слоем катализатора со специально подобранной системой загрузки слоев, состоящих из селективных катализаторов для гидроочистки и гидроконверсии в сочетании с умеренным давлением процесса, что сводит к минимуму образование остаточных продуктов и углеродистых отложений.
Основная схема процесса технологии HIDRO-IMP® включает разделение тяжелой нефти на легкую и тяжелую фракции. Тяжелая фракция подвергается гидроочистке в реакторе с неподвижным слоем, в котором происходит полное удаление металлов и асфальтенов, а также части серы и азота. Частично превращенные продукты из этой стадии вводятся во второй реактор с неподвижным слоем катализатора, где происходит полное превращение сернистых и азотистых органических соединений. Выходящий из реактора поток поступает в сепаратор высокого давления, где жидкие продукты отделяют от газов. Поток жидкости из сепаратора высокого давления подвергается дополнительной очистке, при которой из него удаляется оставшийся растворенный сероводород. Газовую смесь из сепаратора высокого давления подают в блок очистки с целью удаления сероводорода и аммиака, в результате поток чистого водорода повторно сжимают и возвращают в реакционную систему. Такая система позволяет модернизировать процесс нефтепереработки и улучшить качество конечных продуктов.
Завершил пленарную сессию экономический анализ современного состояния отрасли нефтепереработки в России. Лекцию представил ведущий специалист Высшей школы экономики (Москва, Россия), профессор Валерий Анатольевич Крюков: «Нефтяная дилемма России – двигаться на северо-восток или вглубь?»

Основной темой этой лекции стала экономическая основа добычи жидких углеводородов в России. Лекция продемонстрировала основные тенденции в добыче углеводородов, а также шаги и меры, необходимые для развития производства и экспорта продукции.
Россия является одним из крупнейших производителей и экспортеров нефти и газа. Добыча углеводородов играет решающую роль в российской экономике, сектор нефти и газа в России отвечает за 20% ВВП, 52% федерального бюджета (по данным за 2014 год), 67% экспортной выручки, 25% инвестиций в основной капитал.
Разработка открытых месторождений является естественным источником экспорта в западном направлении, в то время как новые месторождения и новые районы, расположенные на северо-востоке России, тяготеют к азиатско-тихоокеанскому рынку. В то же время, северные и центральные арктические месторождения близки как к европейскому, так и к азиатскому рынку, а точный маршрут зависит от транспортных средств и ценовых условий. Важной особенностью развития российской нефтепереработки является то, что места добычи и заводы находятся довольно далеко друг от друга. В то время как основные месторождения расположены в Сибири, большинство нефтеперерабатывающих заводов размещены в европейской части России, на расстоянии более полутора тысяч километров от месторождений. Исторически сложилось, что все трубопроводы были ориентированы с востока на запад. Только два были направлены по-другому: из Западной Сибири на юг (в Казахстан) и на восток (на нефтеперерабатывающий завод в Ангарске, расположенный в Восточной Сибири). В связи с этим возникают вопросы реорганизации промышленности и управления ресурсами. С середины 90-х годов структура сектора коренным образом изменилась по следующим характеристикам: формирование вертикально интегрированных компаний, в том числе для производства, переработки и реализации нефтепродуктов; частичная приватизация вновь созданных компаний; вывод магистральных нефтепроводов из-под контроля государства; предоставление компаниям возможностей для экспорта нефти и нефтепродуктов. Трансформация привела к возрождению добычи нефти в России, основными факторами которого стали доступ к источникам капитала, использование современных технологий, а также возможность распределять ресурсы в соответствии с приоритетами компании.
По оценкам Министерства природных ресурсов, в 2016 году уровень добычи нефти может достигнуть 52 млн тонн в год, однако для этого потребуется дополнительное инвестирование в размере до 100 млрд долларов США. На всех крупных разрабатываемых месторождениях наблюдается спад уровня производства, в то время как недавно обнаруженные месторождения имеют значительно меньшие запасы сырья. Условия добычи нефти также меняются в связи с резким увеличением числа месторождений со сложными характеристиками пласта (что делает проблематичным использование традиционных технологий добычи) и высокими показателями вязкости нефти.

Кроме тяжелой нефти, Россия имеет значительный потенциал в добыче природного сжиженного газа (легких и сверхлегких нефтяных и газовых конденсатов). Производство этих углеводородов связано с разработкой газоконденсатных месторождений в северной части Западной Сибири, в Ямало-Ненецком автономном округе, а также на шельфе арктических морей России. Тем не менее, сам по себе потенциал не означает высокого уровня эффективности производства. Для развития промышленности требуются не только обильные инвестиции, но также и изменение самой структуры промышленности, ориентация на показатели экономической эффективности, развитие технологической базы. В период высоких цен и интенсивной реконструкции существующих месторождений структура нефтяной отрасли стала гораздо более монополизированной, чем раньше (крупные и вертикально-интегрированные компании производят более 85% российской нефти). Динамика добычи жидких углеводородов в России, а также экспорта на внешние рынки в значительной степени будет зависеть от того, насколько хорошо Россия сможет адаптироваться к изменяющимся условиям современного рынка. И решающую роль в этом, несомненно, сыграет развитие технологий и обеспечение свободного доступа к ним.
Участники симпозиума дали высокую оценку научной составляющей симпозиума. Кроме пленарных лекций, программа включала ключевые, устные и стендовые доклады. Несмотря на то, что симпозиум проходил в статусе международного первый раз, в нем приняли участие лидеры в области развития технологий гидрогенизационных процессов нефтепереработки, представлявшие как ведущие университеты мира, так и крупные нефтехимические компании. Это позволило провести мероприятие на высоком международном уровне. Интерес к программе симпозиума позволил привлечь к участию широкий круг экспертов в области разработки и применения новых катализаторов для осуществления гидропроцессов.
Тезисы представленных на форуме докладов можно найти на веб-сайте
Для участников симпозиума была организована экскурсия на нефтеперерабатывающий завод, принадлежащий компании «Нефтяная индустрия Сербии» (НИС), расположенный в городе Панчево. Завод производит весь спектр нефтепродуктов – от моторных бензинов и дизельного топлива до машинных масел и сырья для нефтехимической промышленности. После модернизации, завершившейся в 2010 году, завод производит бензин стандарта Евро-5. Продукция НИС покрывает около 70% розничного топливного рынка Сербии, экспортирует моторные топлива, бензол, толуол, дорожный и промышленный битумы в страны Европейского Союза, а также в соседние страны региона.
Финансовая поддержка мероприятию была оказана Генеральным спонсором симпозиума ПАО «Газпром Нефть», а также ООО «НПК «Синтез» (Барнаул) и ООО «РИОС-Инжиниринг» (Омск). Благодаря этой поддержке организаторы смогли пригласить для представления пленарных и ключевых лекций специалистов мирового уровня из разных стран, дать возможность молодым ученым продемонстрировать результаты своей научной деятельности и пообщаться с мэтрами мирового уровня в области нефтепереработки. Активное взаимодействие с Генеральным спонсором симпозиума ПАО «Газпром Нефть» позволило привлечь к участию в симпозиуме представителей сербской нефтяной индустрии, а также широкий круг студентов и аспирантов университетов Сербии.
Труды симпозиума будут опубликованы в журнале «Катализ в промышленности».
Участники отметили высокий уровень организации симпозиума и его особенную атмосферу, которая позволила им продемонстрировать свой творческий и научный потенциал. На закрытии было принято решение о регулярном проведении Международного Научно-технологического Симпозиума «Нефтепереработка: катализаторы и гидропроцессы» в последующие годы.
Материал подготовили
С.С. Логунова, Т.В. Замулина, А.Н. Загоруйко, О.В. Климов, А.С. Носков
(Институт катализа СО РАН, Новосибирск)
5-6 сентября 2015 г., Казань, Россия
5 и 6 октября в Казанском национальном исследовательском технологическом университете состоялась 4-я молодежная школа-конференция «Каталитический дизайн: от исследований на молекулярном уровне к практической реализации». Это мероприятие проводится Институтом катализа СО РАН уже четвертый раз. Впервые молодежная конференция по катализу была проведена в Новосибирске 2-6 декабря 2002 года. Во второй раз школа-конференция проходила в республике Горный Алтай 25-29 июля 2005 года. Третья школа-конференция состоялась в 2009 году в Свердловской области на базе туристического центра «Чусовая» с 13 по 19 июля. В этом году, несмотря на значительный перерыв, удалось возобновить традицию проведения подобных школ для молодых ученых-каталитиков. Кроме того, эта школа-конференция стала сателлитным симпозиумом XII Европейского конгресса по катализу Europacat-XII, который впервые проводился в России, в городе Казань.
В конференции приняли участие более 80 молодых ученых, студентов, аспирантов и преподавателей вузов из разных городов России (Новосибирск, Казань, Москва, Черноголовка, Тверь, Екатеринбург), стран СНГ (Казахстан и Узбекистан), а также Австрии, Австралии и Франции. В программу конференции были включены 30 устных презентаций различной продолжительности и 43 стендовых доклада молодых ученых. Для чтения пленарных лекций были приглашены ведущие ученые в области катализа из России (Новосибирск, Тверь, Казань), Франции (Страсбург), Мексики (Энсенада), Ирландии (Лимерик), Испании (Жирона), Финляндии (Турку) и Турции (Анкара). Участники прослушали 9 пленарных лекций.
Проведение конференции позволило существенно расширить научные знания молодых ученых и преподавателей в области катализа, а привлечение ведущих российских и мировых специалистов – лидеров в своих областях наук для чтения лекций дало возможность познакомиться с передовыми научными достижениями. Несмотря на небольшую продолжительность школы-конференции, которая включала всего два полных дня работы, научная программа была очень насыщенной. Лекции, устные доклады и постерные презентации охватывали разнообразные тематики, актуальные для гомогенного и гетерогенного катализа, в частности:
 Доктор Мигель Костас Сальгейро
(Университет Жироны, Испания) в своей обзорной лекции рассказал, как созданные природой принципы строения активных центров в ферментах, в частности, содержащих ионы железа и марганца, могут быть использованы при дизайне новых каталитических систем. На примере большого числа работ своей группы лектор убедительно показал, что можно, руководствуясь этими принципами, подобрать необходимый лиганд для комплексов железа или марганца и получить каталитическую систему с высокой селективностью в окислении C-H и C=C связей в различных органических субстратах. В качестве окислителя во всех приведенных реакциях участвовала перекись водорода, что безопасно для окружающей среды и соответствует принципам зеленой химии.
Доктор Мигель Костас Сальгейро
(Университет Жироны, Испания) в своей обзорной лекции рассказал, как созданные природой принципы строения активных центров в ферментах, в частности, содержащих ионы железа и марганца, могут быть использованы при дизайне новых каталитических систем. На примере большого числа работ своей группы лектор убедительно показал, что можно, руководствуясь этими принципами, подобрать необходимый лиганд для комплексов железа или марганца и получить каталитическую систему с высокой селективностью в окислении C-H и C=C связей в различных органических субстратах. В качестве окислителя во всех приведенных реакциях участвовала перекись водорода, что безопасно для окружающей среды и соответствует принципам зеленой химии.
 Профессор Эмрах Озензой
(Университет Бликент, Анкара, Турция) представил увлекательный доклад об особенностях работы катализаторов дожигания выхлопных газов автомобилей. На примере работ, выполненных в его лаборатории методами исследования поверхности, автор продемонстрировал современные подходы к улучшению характеристик катализаторов дожигания. В докладе также обсуждались современные экологические нормы к содержанию вредных веществ в выхлопных газах и технологические новинки, разработанные компанией Toyota Motor. Лекция профессора Озензоя включала в себя большое количество поясняющих видеофрагментов, которые демонстрировали процессы, происходящие в автомобильных катализаторах, и основные принципы работы катализаторов дожигания. Подобный подход значительно облегчил восприятие научного материала и вызвал большой интерес даже у людей, не работающих в данной области.
Профессор Эмрах Озензой
(Университет Бликент, Анкара, Турция) представил увлекательный доклад об особенностях работы катализаторов дожигания выхлопных газов автомобилей. На примере работ, выполненных в его лаборатории методами исследования поверхности, автор продемонстрировал современные подходы к улучшению характеристик катализаторов дожигания. В докладе также обсуждались современные экологические нормы к содержанию вредных веществ в выхлопных газах и технологические новинки, разработанные компанией Toyota Motor. Лекция профессора Озензоя включала в себя большое количество поясняющих видеофрагментов, которые демонстрировали процессы, происходящие в автомобильных катализаторах, и основные принципы работы катализаторов дожигания. Подобный подход значительно облегчил восприятие научного материала и вызвал большой интерес даже у людей, не работающих в данной области.
 Профессор Оксана Анатольевна Холдеева (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск, Россия) рассказала об исследованиях, нацеленных на перевод высокоэффективных гомогенных каталитических систем в гетерогенное состояние. Актуальность этой задачи обусловлена привлекательностью гетерогенных катализаторов для химической и фармацевтической промышленности в связи с более легкой очисткой продуктов после проведения каталитической реакции. Были рассмотрены подходы, применяемые для закрепления активных частиц на разнообразных пористых структурах, а также методы исследования этих систем. Кроме того, О.А. Холдеева рассказала о некоторых «подводных камнях», с которыми может столкнуться исследователь, например, учет возможности вымывания активного компонента в раствор. Интересно было узнать о примерах успешной реализации подобного подхода для получения важных фармацевтических препаратов.
Профессор Оксана Анатольевна Холдеева (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск, Россия) рассказала об исследованиях, нацеленных на перевод высокоэффективных гомогенных каталитических систем в гетерогенное состояние. Актуальность этой задачи обусловлена привлекательностью гетерогенных катализаторов для химической и фармацевтической промышленности в связи с более легкой очисткой продуктов после проведения каталитической реакции. Были рассмотрены подходы, применяемые для закрепления активных частиц на разнообразных пористых структурах, а также методы исследования этих систем. Кроме того, О.А. Холдеева рассказала о некоторых «подводных камнях», с которыми может столкнуться исследователь, например, учет возможности вымывания активного компонента в раствор. Интересно было узнать о примерах успешной реализации подобного подхода для получения важных фармацевтических препаратов.
 В рамках пленарной лекции были представлены результаты многолетней работы группы под руководством Эсфири Михайловны Сульман (Тверской государственный технический университет, Тверь, Россия) по синтезу и исследованию металл-содержащих наночастиц размером 1-3 нм в наноструктурированных полимерах различного типа. Были рассмотрены результаты совместной работы российских и зарубежных исследователей в области создания эффективных каталитических систем на основе наноструктурированных полимеров и металлов Pd, Ru, Cu, Zn для таких каталитических процессов переработки биомассы как гидродеоксигенация, гидролитическая дегидрогенация целлюлозы, синтез метанола в жидкой фазе.
В рамках пленарной лекции были представлены результаты многолетней работы группы под руководством Эсфири Михайловны Сульман (Тверской государственный технический университет, Тверь, Россия) по синтезу и исследованию металл-содержащих наночастиц размером 1-3 нм в наноструктурированных полимерах различного типа. Были рассмотрены результаты совместной работы российских и зарубежных исследователей в области создания эффективных каталитических систем на основе наноструктурированных полимеров и металлов Pd, Ru, Cu, Zn для таких каталитических процессов переработки биомассы как гидродеоксигенация, гидролитическая дегидрогенация целлюлозы, синтез метанола в жидкой фазе.
 Профессор Елена Романовна Савинова
(Университет г. Страсбурга, Франция) подробно и очень доступно рассказала об основных принципах работы и устройстве электрохимической ячейки и электрокатализаторов, о том, чем они отличаются от гетерогенных катализаторов, и какие дополнительные сложности возникают при их исследовании. В частности, было подробно показано, что важно исследовать взаимодействие между катализатором и электролитом и понять, каким именно образом возникает двойной электрический слой. Один из возможных подходов для исследования подобных поверхностных явлений – метод in-situ рентгеновской фотоэлектронной спектроскопии, который позволяет отследить изменения, происходящие на электроде в процессе работы электрохимической ячейки. Были продемонстрированы некоторые результаты подобных исследований для платиновых электрокатализаторов и принципы создания ячеек для in-situ изменений.
Профессор Елена Романовна Савинова
(Университет г. Страсбурга, Франция) подробно и очень доступно рассказала об основных принципах работы и устройстве электрохимической ячейки и электрокатализаторов, о том, чем они отличаются от гетерогенных катализаторов, и какие дополнительные сложности возникают при их исследовании. В частности, было подробно показано, что важно исследовать взаимодействие между катализатором и электролитом и понять, каким именно образом возникает двойной электрический слой. Один из возможных подходов для исследования подобных поверхностных явлений – метод in-situ рентгеновской фотоэлектронной спектроскопии, который позволяет отследить изменения, происходящие на электроде в процессе работы электрохимической ячейки. Были продемонстрированы некоторые результаты подобных исследований для платиновых электрокатализаторов и принципы создания ячеек для in-situ изменений.
 Профессор Дмитрий Юрьевич Мурзин
(Университет Або, Турку, Финляндия) сосредоточил внимание аудитории на проблемах переработки в полезные продукты наиболее сложных, с точки зрения химии, источников углеводородов – биомассы древесины, а также остаточных жиров растительного и животного происхождения. Были представлены различные способы переработки древесины, от биодизеля до чистых химических продуктов, масел и строительных материалов. Основные подходы к переработке этого сложного сырья профессор Мурзин рассмотрел на примере существующих промышленных объектов, которые применяют те или иные процессы. В качестве основных методов были отмечены: химическое и каталитическое фракционирование древесины, термический или каталитический пиролиз биомассы для получения синтез газа или биотоплив, гетеро-каталитическая деполимеризации целлюлозы и лигнина с получением низкомолекулярных продуктов – строительных блоков для синтеза широкого спектра полезных продуктов. Отдельно были отмечены каталитические процессы выделения ценных химических веществ из природной древесины. В конце лекции было выделено несколько типов важных реакций, для которых еще не существует катализатора, и предложено всем молодым участникам направить свои усилия на решение этой проблемы.
Профессор Дмитрий Юрьевич Мурзин
(Университет Або, Турку, Финляндия) сосредоточил внимание аудитории на проблемах переработки в полезные продукты наиболее сложных, с точки зрения химии, источников углеводородов – биомассы древесины, а также остаточных жиров растительного и животного происхождения. Были представлены различные способы переработки древесины, от биодизеля до чистых химических продуктов, масел и строительных материалов. Основные подходы к переработке этого сложного сырья профессор Мурзин рассмотрел на примере существующих промышленных объектов, которые применяют те или иные процессы. В качестве основных методов были отмечены: химическое и каталитическое фракционирование древесины, термический или каталитический пиролиз биомассы для получения синтез газа или биотоплив, гетеро-каталитическая деполимеризации целлюлозы и лигнина с получением низкомолекулярных продуктов – строительных блоков для синтеза широкого спектра полезных продуктов. Отдельно были отмечены каталитические процессы выделения ценных химических веществ из природной древесины. В конце лекции было выделено несколько типов важных реакций, для которых еще не существует катализатора, и предложено всем молодым участникам направить свои усилия на решение этой проблемы.
 Артем Николаевич Безруков (КНИТУ, Казань, Россия) представил обзорную лекцию, посвященную каталитическим исследованиям, проводимым в Казанском национальном исследовательском технологическом университете (КНИТУ). В большинстве случаев эти исследования имеют прикладную направленность, и многие результаты используются на нефтехимических предприятиях республики Татарстан. Так, например, были разработаны и внедрены на заводе «Нижнекамскнефтехим» различные каталитические системы для окисления этилбензола, легких парафинов и топливных сульфидов. Активно исследуются катализаторы полимеризации олефинов, синтеза и регенерации отработанных катализаторов в сверхкритических условиях, а также системы для переработки тяжелых нефтей. Кроме того, подробно были представлены результаты теоретических исследований возможных механизмов протекания реакции разложения высокоэнергетических соединений с использованием методов квантовой химии и молекулярной динамики.
Артем Николаевич Безруков (КНИТУ, Казань, Россия) представил обзорную лекцию, посвященную каталитическим исследованиям, проводимым в Казанском национальном исследовательском технологическом университете (КНИТУ). В большинстве случаев эти исследования имеют прикладную направленность, и многие результаты используются на нефтехимических предприятиях республики Татарстан. Так, например, были разработаны и внедрены на заводе «Нижнекамскнефтехим» различные каталитические системы для окисления этилбензола, легких парафинов и топливных сульфидов. Активно исследуются катализаторы полимеризации олефинов, синтеза и регенерации отработанных катализаторов в сверхкритических условиях, а также системы для переработки тяжелых нефтей. Кроме того, подробно были представлены результаты теоретических исследований возможных механизмов протекания реакции разложения высокоэнергетических соединений с использованием методов квантовой химии и молекулярной динамики.
 Профессор Андрей Валерьевич Симаков (Центр нанонауки и нанотехнологии, UNAM, Энсенада, Мексика) прочел популярную лекцию о перспективах применения нанореакторов для более эффективного использования катализаторов относительно массы перерабатываемых субстратов. Были описаны нанореакторы на основе полых, частично открытых сфер размером от 1 до 100 нанометров с различными активными центрами, расположенными во внутренней полости. Такие структуры, как было показано, обладают преимуществами микро и мезопористых катализаторов, но при этом не образуют крупных упорядоченных структур и, как следствие, характеризуются более эффективным транспортом реагентов к активным центрам. Были охарактеризованы основные типы структур и методы создания активных центров внутри присущих им полостей. В частности, лектор рассказал о последних достижениях в области создания нанореакторов, состоящих из полых сфер оксида циркония с золотыми наночастицами внутри, выступающими в качестве активных центров. Последняя часть доклада была посвящена синтетическим проблемам в создании нанореакторов: уменьшению толщины стенок нанополости при сохранении их стабильности, а также более точному контролю за размером и составом активных центров, создаваемых внутри нанореактора.
Профессор Андрей Валерьевич Симаков (Центр нанонауки и нанотехнологии, UNAM, Энсенада, Мексика) прочел популярную лекцию о перспективах применения нанореакторов для более эффективного использования катализаторов относительно массы перерабатываемых субстратов. Были описаны нанореакторы на основе полых, частично открытых сфер размером от 1 до 100 нанометров с различными активными центрами, расположенными во внутренней полости. Такие структуры, как было показано, обладают преимуществами микро и мезопористых катализаторов, но при этом не образуют крупных упорядоченных структур и, как следствие, характеризуются более эффективным транспортом реагентов к активным центрам. Были охарактеризованы основные типы структур и методы создания активных центров внутри присущих им полостей. В частности, лектор рассказал о последних достижениях в области создания нанореакторов, состоящих из полых сфер оксида циркония с золотыми наночастицами внутри, выступающими в качестве активных центров. Последняя часть доклада была посвящена синтетическим проблемам в создании нанореакторов: уменьшению толщины стенок нанополости при сохранении их стабильности, а также более точному контролю за размером и составом активных центров, создаваемых внутри нанореактора.
 Профессор Сергей Александрович Белошапкин (Институт материалов и поверхности, Университет г. Лимерика, Ирландия) рассмотрел в своей лекции основы метода время-пролетной вторичной ионной масс-спектрометрии (ВИМС) и особенности применения данного метода для исследования свойств катализаторов. Благодаря высокой поверхностной чувствительности и возможности получить детальную информацию об элементном и молекулярном составе поверхности, данный метод в последнее годы становится все более популярным для исследования поверхности гетерогенных катализаторов. Автор показал технологические особенности масс-спектрометрических приборов и проблемы, с которыми может столкнуться пользователь, такими как сложность интерпретации масс-спектров и получения количественных характеристик. Также профессор Белошапкин продемонстрировал возможности использования метода ВИМС для исследования сложных каталитических систем на основе смешанных оксидов и анализа процессов формирования гетерогенных катализаторов из металл-органических предшественников.
Профессор Сергей Александрович Белошапкин (Институт материалов и поверхности, Университет г. Лимерика, Ирландия) рассмотрел в своей лекции основы метода время-пролетной вторичной ионной масс-спектрометрии (ВИМС) и особенности применения данного метода для исследования свойств катализаторов. Благодаря высокой поверхностной чувствительности и возможности получить детальную информацию об элементном и молекулярном составе поверхности, данный метод в последнее годы становится все более популярным для исследования поверхности гетерогенных катализаторов. Автор показал технологические особенности масс-спектрометрических приборов и проблемы, с которыми может столкнуться пользователь, такими как сложность интерпретации масс-спектров и получения количественных характеристик. Также профессор Белошапкин продемонстрировал возможности использования метода ВИМС для исследования сложных каталитических систем на основе смешанных оксидов и анализа процессов формирования гетерогенных катализаторов из металл-органических предшественников.
 Много докладов молодых ученых было посвящено проблемам приготовления катализаторов. Так, А.П. Коскин (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск, Россия) представил доклад о разработке новых кислотных катализаторов на основе углеродных материалов. Е.В. Бессуднова (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск, Россия) рассказала о приготовлении наноразмерного диоксида титана методом золь-гель химии.
Много докладов молодых ученых было посвящено проблемам приготовления катализаторов. Так, А.П. Коскин (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск, Россия) представил доклад о разработке новых кислотных катализаторов на основе углеродных материалов. Е.В. Бессуднова (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск, Россия) рассказала о приготовлении наноразмерного диоксида титана методом золь-гель химии.  Очень впечатляющий доклад, с интересными иллюстрациями и слайдами, был представлен Хамидом Арандьяном
(Университет Южного Уэльса, Сидней, Австралия). Темой доклада было получение новых перовскитных материалов высокой пористости методом темплатного синтеза. Несмотря на то, что данный подход достаточно хорошо известен, для перовскитных систем он применяется не так часто, однако имеет большие перспективы, как это было показано в докладе доктора Арандьяна.
Очень впечатляющий доклад, с интересными иллюстрациями и слайдами, был представлен Хамидом Арандьяном
(Университет Южного Уэльса, Сидней, Австралия). Темой доклада было получение новых перовскитных материалов высокой пористости методом темплатного синтеза. Несмотря на то, что данный подход достаточно хорошо известен, для перовскитных систем он применяется не так часто, однако имеет большие перспективы, как это было показано в докладе доктора Арандьяна.
 Особый интерес участников вызвал доклад Е.Ю. Асалиевой
(Технологический институт сверхтвердых и новых углеродных материалов, Москва, Россия), посвященный приготовлению новых композитных катализаторов для синтеза Фишера-Тропша на основе никеля Ренея. По итогам конференции этот доклад был признан одним из лучших устных докладов. Интересный доклад, посвященный исследованию динамики и подвижности молекул углеводородов в порах
Особый интерес участников вызвал доклад Е.Ю. Асалиевой
(Технологический институт сверхтвердых и новых углеродных материалов, Москва, Россия), посвященный приготовлению новых композитных катализаторов для синтеза Фишера-Тропша на основе никеля Ренея. По итогам конференции этот доклад был признан одним из лучших устных докладов. Интересный доклад, посвященный исследованию динамики и подвижности молекул углеводородов в порах  цеолитов, представил Д.И. Колоколов (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск, Россия). Основной метод, применяемый для подобных исследований — твердотельная ЯМР спектроскопия на ядрах дейтерия. Несмотря на сложность и уникальность подобного метода исследования, автору удалось доступно показать его возможности, а также продемонстрировать полученные результаты на конкретных примерах.
цеолитов, представил Д.И. Колоколов (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск, Россия). Основной метод, применяемый для подобных исследований — твердотельная ЯМР спектроскопия на ядрах дейтерия. Несмотря на сложность и уникальность подобного метода исследования, автору удалось доступно показать его возможности, а также продемонстрировать полученные результаты на конкретных примерах.
 Модельным исследованиям поверхности методом послойного напыления в системах Pd-ZrO2
и Cu-ZrO2
был посвящен доклад аспиранта из Университета г. Инсбрук (Австрия) Лукаса Майера. Автором было показано, что при напылении подобных систем в различной последовательности можно детально разобраться в механизме окислительных превращений CO, углекислого газа и метана. Научный руководитель Лукаса — доктор Симон Пеннер также выступил с устным докладом, рассказав об исследовании перовскитных катализаторов в реакциях сдвига водяного пара и окисления метана. Были продемонстрированы возможности современных физико-химических методов исследования, в частности, просвечивающей электронной микроскопии высокого разрешения.
Модельным исследованиям поверхности методом послойного напыления в системах Pd-ZrO2
и Cu-ZrO2
был посвящен доклад аспиранта из Университета г. Инсбрук (Австрия) Лукаса Майера. Автором было показано, что при напылении подобных систем в различной последовательности можно детально разобраться в механизме окислительных превращений CO, углекислого газа и метана. Научный руководитель Лукаса — доктор Симон Пеннер также выступил с устным докладом, рассказав об исследовании перовскитных катализаторов в реакциях сдвига водяного пара и окисления метана. Были продемонстрированы возможности современных физико-химических методов исследования, в частности, просвечивающей электронной микроскопии высокого разрешения.
 Доклад Д.В. Красникова (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск, Россия) был посвящен актуальной тематике — получению углеродных материалов, в частности, многослойных углеродных нанотрубок. Были представлены результаты исследования процесса активации металлических катализаторов роста нанотрубок. Показано, что для понимания всех деталей процесса на атомном уровне необходимо применение нескольких комплементарных методов физико-химического и структурного исследования.
Доклад Д.В. Красникова (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск, Россия) был посвящен актуальной тематике — получению углеродных материалов, в частности, многослойных углеродных нанотрубок. Были представлены результаты исследования процесса активации металлических катализаторов роста нанотрубок. Показано, что для понимания всех деталей процесса на атомном уровне необходимо применение нескольких комплементарных методов физико-химического и структурного исследования.  Методам in-situ исследования процессов получения карбидов и графена на никелевых модельных системах был посвящен очень интересный доклад Рафаэля Рамешана (Университет г. Инсбрук, Австрия).
Методам in-situ исследования процессов получения карбидов и графена на никелевых модельных системах был посвящен очень интересный доклад Рафаэля Рамешана (Университет г. Инсбрук, Австрия).
 Доклады участников продемонстрировали все увеличивающуюся актуальность исследования каталитических систем переработки и облагораживания растительного сырья для получения важных химических продуктов и топлива. Так, проблемам переработки продуктов производства бионефти – этанола и глицерола – был посвящен доклад М.В. Араповой (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск, Россия). Н.В. Громов рассказал о проблемах переработки целлюлозы, а
Доклады участников продемонстрировали все увеличивающуюся актуальность исследования каталитических систем переработки и облагораживания растительного сырья для получения важных химических продуктов и топлива. Так, проблемам переработки продуктов производства бионефти – этанола и глицерола – был посвящен доклад М.В. Араповой (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск, Россия). Н.В. Громов рассказал о проблемах переработки целлюлозы, а
Обсуждение постерных докладов проходило очень оживленно, вызывая большой интерес участников. Особо были отмечены доклады Е.Е. Файнгольда и И.В. Жаркова (Институт проблем химической физики РАН, Черноголовка, Россия), посвященные синтезу, исследованию и применению акрилоксиизобутилалюминиевых соединений для активации металлоценовых комплексов полимеризации олефинов. Доклад Е. Файнгольда был выделен как один из лучших постерных докладов на школе-конференции.
Молодые ученые отметили высокий уровень проведения школы-конференции, актуальность приглашенных докладов и изъявили желание участвовать в последующих школах-конференциях, организуемых Институтом катализа СО РАН. Важность продолжения традиции проведения подобных научно-образовательных мероприятий подчеркнул руководитель группы аспирантов и постдоков из университета города Инсбрук, доктор Симон Пеннер, поскольку в 2002 году он сам, будучи аспирантом, участвовал в первой молодежной школе-конференции по катализу, проходившей в Новосибирске.

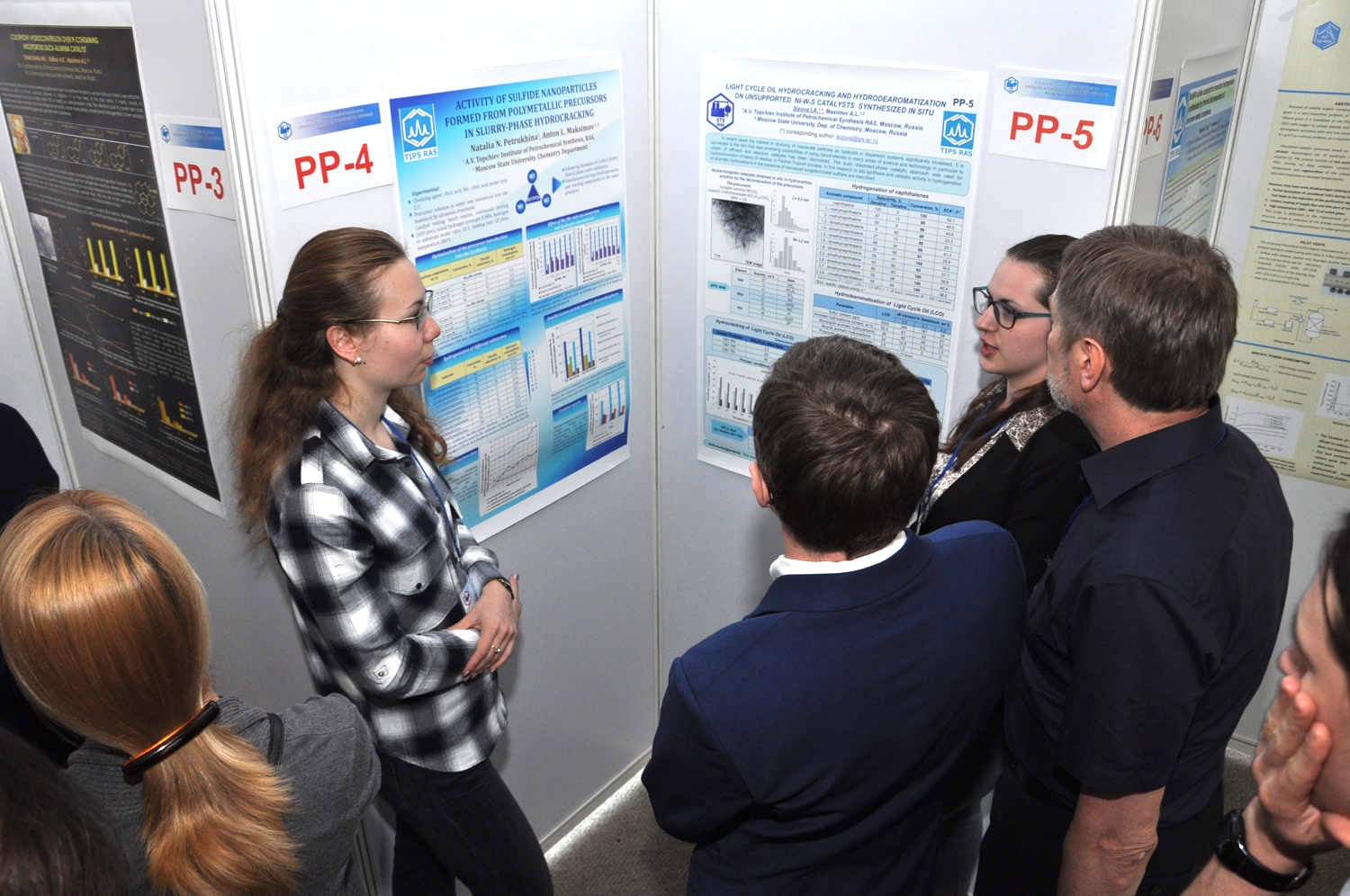
Supposedly unreactive ligand caught in the act of protonation, suggesting new opportunities in catalyst design
Discovery of the rhodium Cp*H intermediate has revealed a new type of metal-ligand cooperation that could benefit catalyst design.
Sometimes chemists set themselves up for a surprise. Following sets of experiments in which something doesn’t happen and doesn’t seem likely to happen, they soon believe it never will. Until it does.
Two research groups have independently uncovered one of these surprises involving the popular transition-metal ligand pentamethylcyclopentadienyl, or Cp*.
Chemists have traditionally thought of cyclopentadienyl ligands as being “innocent,” which means they offer electronic support to a metal catalyst but generally don’t do anything chemically. The two groups were studying reactions involving Cp*Rh(bipyridine), often used in hydrogenation reactions and in hydrogen-forming reactions, when they found that the expected metal hydride intermediate was followed by formation of an unexpected intermediate in which the hydrogen had migrated to one of the carbon atoms in the Cp* ring.
“These two reports showing that the seemingly innocent Cp* ligand can reversibly form a C–H bond by proton transfer from rhodium hydride are remarkable,” comments chemistry professor David Milstein of the Weizmann Institute of Science, who was not involved in the research. “Considering the ubiquity of cyclopentadienyl metal complexes in homogeneous catalysis, this pathway should be seriously considered in the design and understanding of reactions in which proton/hydride transfer may be involved.”
Alexander J. M. Miller of the University of North Carolina, Chapel Hill, who led one of the teams, says chemists had previously worked out mechanisms involving hydride intermediates that made sense and thought the story ended there. But they did not exercise due diligence and poke around enough to see that a protonated Cp* intermediate, denoted Cp*H, could be involved as well. “What’s more surprising,” Miller points out, “the Cp*H complex is not a dead end. This diene complex is still an active catalyst.”
Miller’s group came across the Cp*H intermediate while investigating hydride transfer reactions with the cellular enzyme cofactor nicotinamide adenine dinucleotide (NAD+) to form the reduced product NADH (Chem. Commun. 2016, DOI: 10.1039/c6cc00575f).
Meanwhile, a team led by Harry B. Gray and Jay R. Winkler at Caltech and James D. Blakemore at the University of Kansas discovered the Cp*H intermediate while investigating the coupling of protons to form H2 when treating Cp*Rh(bipyridine) with acid (Proc. Natl. Acad. Sci. USA 2016, DOI: 10.1073/pnas.1606018113).
“These discoveries illustrate the versatility of mechanisms by which protons and hydrides can be delivered to and from metals,” comments Morris Bullock, director of the Center for Molecular Electrocatalysis at Pacific Northwest National Laboratory. “While these examples are for rhodium, the prevalence of cyclopentadienyl ligands in organometallic catalysts raises the possibility that similar reactivity could be widespread and involve other metals, and may be intentionally exploited in the design of new catalysts.”
Elements 113, 115, 117, and 118 are likely to become nihonium, moscovium, tennessine, and oganesson
Nihonium, moscovium, tennessine, and oganesson are the recommended names for elements 113, 115, 117, and 118, the International Union of Pure & Applied Chemistry (IUPAC) announced today.
IUPAC officially added the elements to the periodic table at the end of 2015. Those credited with discovering the elements get the rights to propose permanent names and symbols. The names will be finalized after public review and formal approval by the IUPAC Council.

The proposed names for elements 113, 115, 117, and 118 are nihonium, moscovium, tennessine, and oganesson.
According to recommendations published by IUPAC in April, elements can be named after a mythological concept, a mineral, a place or country, a property, or a scientist (Pure Appl. Chem.2016, DOI: 10.1515/pac-2015-0802). Element names should also “have an ending that reflects and maintains historical and chemical consistency,” the recommendations say. “This would be in general ‘-ium’ for elements belonging to groups 1–16, i.e. including the f-block elements, ‘-ine’ for elements of group 17 and ‘-on’ for elements of group 18.”
Japan’s RIKEN research institution was credited with discovering element 113. Nihonium (Nh) comes from Nihon, which is one of two ways to say “Japan” in Japanese. It is the first element discovered in and named after an Asian country.
The discoveries of the other three elements were credited to European-American collaborations involving Russia’s Joint Institute for Nuclear Research and the U.S. Lawrence Livermore and Oak Ridge national laboratories.
Moscovium (Mc) recognizes Moscow and its surrounding area “and honors the ancient Russian land that is the home of the Joint Institute for Nuclear Research,” says an IUPAC press release.
Tennessine (Ts) “is in recognition of the contribution of the Tennessee region, including Oak Ridge National Laboratory, Vanderbilt University, and the University of Tennessee at Knoxville, to superheavy element research, including the production and chemical separation of unique actinide target materials for superheavy element synthesis,” the release also says.
Oganesson (Og) honors Russian nuclear physicist Yuri T. Oganessian, who leads the Flerov Laboratory of Nuclear Reactions at the Joint Institute for Nuclear Research. He joins his countrymen Vasili Samarsky-Bykhovets (samarium), Dmitri Mendeleev (mendelevium), and Georgy Flerov (flerovium) in having a namesake element.
In April, Nature Chemistry published element name predictions by freelance writer Philip Ball, Worcester Polytechnic Institute chemistry professor Shawn Burdette, chemistry writer and blogger Kat Day, UCLA lecturer and author Eric Scerri, and Stockholm University chemistry researcher Brett Thornton (Nat. Chem. 2016, DOI: 10.1038/nchem.2482).
Burdette came closest to the proposed names, suggesting nipponium (Nippon is the other way to say “Japan” in Japanese), moscovium, tennessine, and oganesson. “After my element prediction success, I’m retiring from chemistry,” he joked on Twitter. “I can’t see my career going any higher.”
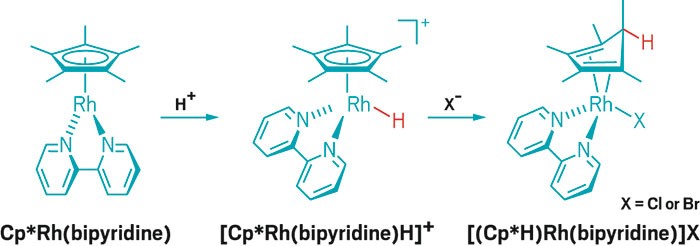
Chalcogel catalyst reduces dinitrogen in water under ambient temperature and pressure
To produce the large quantities of fertilizer needed to sustain global food production, industry relies on the Haber-Bosch process to split dinitrogen from the air to synthesize ammonia. But to generate the high temperatures and pressures needed for the reaction, ammonia plants consume fossil fuels and, as a result, contribute significantly to greenhouse gas emissions.
 This chalcogel, a network of Fe4S4 clusters linked with [Sn2S6]4– anions, uses visible light to convert N2 to NH3.
This chalcogel, a network of Fe4S4 clusters linked with [Sn2S6]4– anions, uses visible light to convert N2 to NH3.
Researchers want to develop a green alternative to the Haber-Bosch process. In a new effort, Mercouri G. Kanatzidis and George C. Schatz of Northwestern University and coworkers used iron-sulfur (Fe4S4) clusters, found naturally in the nitrogenase enzymes bacteria use to split dinitrogen, to make a light-driven catalyst that converts N2 to NH3 in water at ambient temperature and pressure (Proc. Natl. Acad. Sci. USA 2016, DOI: 10.1073/pnas.1605512113).
After synthesizing the Fe4S4 clusters, the researchers link them with [Sn2S6]4– anions and form the complex into a chalcogel, a foamy gel containing the chalcogen element sulfur. The black material absorbs visible light and uses that energy to break the N≡N triple bond, one of the strongest in chemistry. The nitrogen atoms then combine with water-derived hydrogen to make NH3.
Other researchers have developed transition metal-N2 complexes that convert N2 to NH3, but those systems typically require organic solvents, low temperatures, and/or high pressures. Some previously developed light-based systems also can reduce N2, but the chalcogel is 10 times as efficient.
Hybrid synthetic-biological approaches, such as a nanorod-nitrogenase system (Science 2016, DOI: 10.1126/science.aaf2091, C&EN, April 25, page 9), are much more efficient than the chalcogel system. But the hybrid approach has limitations: Obtaining nitrogenase from organisms is difficult, and the enzymes are not stable for more than a few hours after isolation, Kanatzidis says.
The chalcogel is orders of magnitude less efficient than the Haber-Bosch process. But unlike the Haber-Bosch process or metal-N2 complexes, the chalcogel catalyst can fix N2 under quite mild conditions using light as the only driving force, comments photocatalysis specialist Lizhi Zhang of Central China Normal University. Understanding of the chalcogel mechanism remains fragmentary at this point, he says, so further study is still needed to find ways to optimize the catalyst.
Catalysis expert Patrick Holland of Yale University says it isn’t fair to compare the efficiency of the chalcogel to the Haber-Bosch method because the industrial process has been optimized for more than a century. “The chalcogel already makes reasonable amounts of ammonia, so it’s a valid first step toward using light to drive N2 reduction,” he says. “The catalyst is very modifiable, so it could in principle be improved a lot in the long run.”
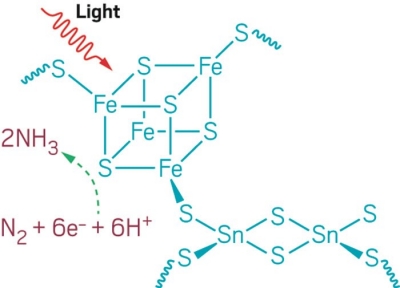
Simulations suggest that carbon’s one-dimensional allotrope remains a theory
Sixteen days before his death, Nobel Laureate Harold W. Kroto published a study with colleagues at Arizona State University that ends thusly: “The bottom line is that carbyne, the linear carbon allotrope, remains as elusive and theoretical as ever.”
 A crystal structure of gold-stabilized carbon chains proposed by Kroto, Buseck, and Tarakeshwar. The box shows the unit cell of the “pseudocarbyne.”
A crystal structure of gold-stabilized carbon chains proposed by Kroto, Buseck, and Tarakeshwar. The box shows the unit cell of the “pseudocarbyne.”
The team, led by Peter R. Buseck and Pilarisetty Tarakeshwar of Arizona State, used density functional theory to perform simulations that suggest a recent report of carbyne creation overlooks an important element, namely gold.
Last October, Guowei Yang of Sun Yat-sen University and coworkers reported that they had made carbyne crystals with the help of gold (Sci. Adv. 2015, DOI: 10.1126/sciadv.1500857). Firing a laser at a gold target in ethanol forms a small plasma above the target where carbyne can form, the team said.
Although transmission electron microscopy revealed that gold nanoparticles rested on the exterior of the crystal structures, further analyses, including Raman spectroscopy, led Yang’s team to conclude that gold was not integrated into the crystals. In other words, the substance was carbyne, a string of carbon atoms bonded together with alternating single and triple bonds.
The Arizona State researchers and Kroto contested the report with a letter to Science Advancesin January and now have data from simulations that indicate that the material is composed of carbon chains stabilized by gold nanoparticles (J. Phys. Chem. Lett. 2016, DOI: 10.1021/acs.jpclett.6b00671). The misidentification of gold-stabilized pseudocarbyne as pure carbyne can be chalked up to misinterpreted data, Buseck says.
For example, Yang’s team stated that a peak in an experimental Raman spectrum corresponded to carbon-carbon single bonds in carbyne. Yet the simulations did not reproduce this peak unless linear carbon chains were attached to gold particles. The appearance of this peak is likely due to the same plasmonic effect that metals provide in surface-enhanced Raman spectroscopy, Tarakeshwar says.
Yang stands by his team’s initial assessment, however. Different metal targets, such as palladium, produced crystals that yielded identical spectra, he says, although this was not shown in the October study. Different metals would have different spectra were they part of the crystal. Yang says the computational work is “just a theoretical simulation, not supported by any experimental observations.”
Experimental measurements do exist, but all the data were collected by Yang’s team, Buseck states. He adds that he has unsuccessfully tried to obtain samples from Yang to analyze. Although metal-stabilized carbon chains are not carbyne, they could be an interesting new class of materials, but that’s just speculation until there is more experimental evidence, Buseck says.
Rik R. Tykwinski, who studies long carbon chains at Friedrich Alexander University, Erlangen-Nuremberg, says he fully believes the work done by Kroto, Buseck, and Tarakeshwar. “Kroto often provided the voice of common sense in this field, sometimes quietly and sometimes quite vocally,” Tykwinski says. With this last publication, he adds, “Kroto’s legacy lives on.”
Supported indium oxide catalyst could boost lab-scale process to an industrial level
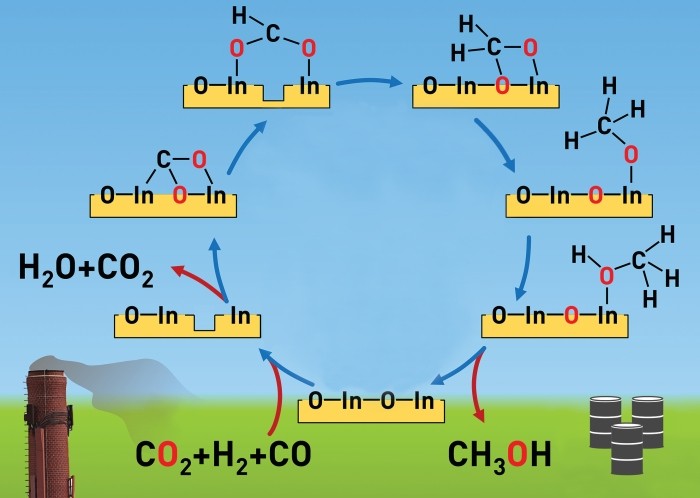
Vacancies on the surface of a ZrO2-supported In2O3 catalyst play a key role in converting CO2 to CH3OH.
Manufacturers generally produce methanol, a key chemical building block and fuel, from petroleum-derived syngas, a mixture of carbon monoxide and hydrogen. Direct hydrogenation of the greenhouse gas carbon dioxide would be a more efficient and environmentally sustainable route to methanol. But practical catalysts capable of making this reaction happen on an industrial scale have been unavailable.
Scientists had shown earlier that indium oxide catalyzes the direct hydrogenation of CO2 to CH3OH on a lab scale. Javier Pérez-Ramírez of ETH Zurich and coworkers now demonstrate that zirconium oxide-supported In2O3 catalyzes the process under conditions similar to those required for industrial production (Angew. Chem. Int. Ed. 2016, DOI: 10.1002/anie.201600943).
The supported catalyst can convert CO2 and H2 to CH3OH over at least 1,000 hours of continuous use and outperforms most other hydrogenation catalysts. The researchers proved experimentally that oxygen vacancies on the catalyst surface make the reaction possible—a mechanism predicted by theoretical calculations from a team led by Qingfeng Ge of Southern Illinois University and Tianjin University (ACS Catal. 2013, DOI: 10.1021/cs400132a).
The ETH Zurich group optimized the reaction by adding CO to the starting materials and varying the temperature, both of which tuned the number of vacancies. The technique is “a long-sought breakthrough with the potential to realize continuous CO2 conversion to methanol on a commercial scale,” Ge says.
Pérez-Ramírez and coworkers have filed patent applications on the technology in collaboration with French energy firm Total, which has started pilot studies of the process.
Chemical & Engineering News
III Российский конгресс по катализу “Роскатализ-2017” будет проведен с 22 по 26 мая 2017 года в Нижнем Новгороде. Предметами для обсуждения участников конгресса станут: современное состояние дел и перспективы развития во всех областях катализа, поиск новых возможностей для импортозамещения в отношении стратегически важных катализаторов и химической продукции, подготовка высококвалифицированных кадров, создание более тесной консолидации отечественной науки, бизнеса и высшего образования для решения фундаментальных и прикладных задач катализа.
ОРГАНИЗАТОРЫ КОНГРЕССА
При поддержке НП “Национальное каталитическое общество”
|
НАУЧНЫЙ КОМИТЕТ КОНГРЕССА |
|
|
Председатели: |
|
| В.И. Бухтияров, чл.-корр. РАН | ИК СО РАН, Новосибирск |
| В.Н. Пармон, академик РАН | ИК СО РАН, Новосибирск |
| Е.В. Чупрунов, д.ф.-м.н. | ННГУ, Нижний Новгород |
| В.В. Лунин, академик РАН | МГУ, Москва |
| С.Н. Хаджиев, академик РАН | ИНХС, Москва |
| М.П. Егоров, академик РАН | ИОХ РАН, Москва |
|
Члены Научного комитета: |
|
| С.М. Алдошин, академик РАН | Президиум РАН, Москва |
| А.А. Берлин, академик РАН | ИХФ РАН, Москва |
| С.С. Галибеев, д.х.н. | ПАО “Сибур Холдинг”, Москва |
| А.Г. Дедов, чл.-корр. РАН | РГУ нефти и газа, Москва |
| У.М. Джемилев, чл.-корр. РАН | Президиум УНЦ РАН, Уфа |
| З.Р. Исмагилов, чл.-корр. РАН | ИУХМ СО РАН, Кемерово |
| С.В. Калюжный, д.х.н. | ОАО “РОСНАНО”, Москва |
| В.М. Капустин, д.т.н. | ОАО “ВНИПИНефть”, Москва |
| Б.Н. Кузнецов, д.х.н. | ИХХТ СО РАН, Красноярск |
| В. Лавренов, к.х.н. | ИППУ СО РАН, Омск |
| В.А. Лихолобов, чл.-корр. РАН | Президиум ОНЦ СО РАН, Омск |
| А.Л. Максимов, д.х.н. | ИНХС РАН, Москва |
| О.Г. Синяшин, академик РАН | ИОФХ РАН, Казань |
| А.Ю. Стахеев, д.х.н. | ИОХ РАН, Москва |
| П.А. Стороженко, чл.-корр. РАН | “ГНИИХТЭОС”, Москва |
| А.Б. Ярославцев, чл.-корр. РАН | ИНХС РАН, Москва |
|
ПРОГРАММНЫЙ КОМИТЕТ |
|
|
Председатели: |
|
| В.А. Яковлев, д.х.н. | ИК СО РАН, Новосибирск |
| А.С. Носков, д.т.н. | ИК СО РАН, Новосибирск |
| Е.В. Сулейманов, д.х.н. | НИИ химии, ННГУ, Н. Новгород |
| Е.С. Локтева, д.х.н. | МГУ, Москва |
|
Члены Программного комитета: |
|
| Л.А. Исупова, д.х.н. | ИК СО РАН, Новосибирск |
| А.В. Князев, д.х.н. | НИИ Химии, ННГУ, Н. Новгород |
| О.П. Таран, д.х.н. | ИК СО РАН, Новосибирск |
| В.В. Каичев, к.ф.-м.н. | ИК СО РАН, Новосибирск |
| Е.А. Козлова, к.х.н. | ИК СО РАН, Новосибирск |
| Д.А. Шляпин, к.х.н. | ИППУ СО РАН, Омск |
| И.В. Седов, к.х.н. | ИПФХ РАН, Черноголовка |
| М.В. Быкова, к.х.н. | ИК СО РАН, Новосибирск |
| Л.С. Кибис, к.х.н. | ИК СО РАН, Новосибирск |
| О.О. Мироненко, к.х.н. | ИК СО РАН, Новосибирск |
|
СЕКРЕТАРИАТ |
|
| М.А. Клюса | ИК СО РАН, Новосибирск |
| О.В. Крашенинникова | ННГУ, Нижний Новгород |
| М.С. Суворова | ИК СО РАН, Новосибирск |
| С.С. Логунова | ИК СО РАН, Новосибирск |
| А.М. Ершова | ИК СО РАН, Новосибирск |
НАУЧНАЯ ПРОГРАММА
Научная программа конгресса “Роскатализ-2017” будет включать пленарные лекции (40 минут), ключевые доклады (30 минут), устные доклады (20 и 10 минут), флеш анонсы постеров молодых учёных (5 минут), стендовые доклады по следующим научным направлениям:
Секция 1. Физико-химические основы катализа
Секция 2. Научные основы производства катализаторов
Секция 3. Перспективные каталитические процессы
Секция 4. Промышленные катализаторы и каталитические процессы
САТЕЛЛИТНЫЕ МЕРОПРИЯТИЯ
Круглый стол I. “Образование и катализ”
Круглый стол II. “Катализ: проблемы импортозамещения.
Процессы, катализаторы, приборное обеспечение”
Симпозиум. “Каталитические процессы в газохимии”
Молодёжная школа по катализу
“Физико-химические методы исследования – ключ
к пониманию принципов каталитического действия”
Основные направления школы:
ПУБЛИКАЦИЯ ТРУДОВ
Сборник всех принятых докладов будет издан в электронном виде с присвоением международного стандартного книжного номера (ISBN). Организационный комитет приглашает участников, выступающих на конгрессе с пленарными лекциями, ключевыми, устными и стендовыми докладами, опубликовать статьи на основе презентационных материалов в номерах журналов “Кинетика и катализ” (доклады секций “Физико-химические основы каталитических процессов” и “Научные основы производства катализаторов”), “Катализ в промышленности” (доклады секций “Перспективные каталитические процессы” и “Промышленные катализаторы и каталитические процессы”), “Альтернативная энергетика и экология”.
ЗАЯВКИ НА УЧАСТИЕ И ТЕЗИСЫ ДОКЛАДОВ
Желающим принять участие в работе конгресса “Роскатализ-2017” необходимо зарегистрироваться на сайте конгресса: http://conf.nsc.ru/ruscatalysis-2017.
КЛЮЧЕВЫЕ ДАТЫ
|
10.06.2016 – 20.10.2016 |
Регистрация участников и подача тезисов докладов |
|
01.12.2016 |
Рассылка уведомления о принятии докладов |
|
28.02.2017 |
Размещение предварительной научной программы на сайте |
|
22.05 2017 – 26.05.2017 |
Рабочие дни |
|
27.05.2017 |
Пост-тур |
КОНТАКТЫ
|
В Новосибирске: |
В Нижнем Новгороде: |
|
Институт катализа |
Нижегородский |
|
August 28 – September 2, 2016 |
|
|
September 4-7, 2016 |
|
|
September 4-8, 2016 |
|
|
4-8 сентября 2016 г. |
|
|
September 5-7, 2016 |
http://catalysis.ru/resources/science/conferences/ |
|
September 5-8, 2016 |
|
|
6-9 сентября 2016 г. |
|
|
7-10 сентября 2016 г. |
|
|
12-15 сентября 2016 г. |
|
|
13-16 сентября 2016 г. |
|
|
15-16 сентября 2016 г. |
admin@ipcet.ru |
|
19-20 сентября 2016 г. |
|
|
September 19-22, 2016 |
http://www.european-mrs.com/meetings/2016-fall |
|
September 19-23, 2016 |
|
|
19-23 сентября 2016 г. |
|
|
19-30 сентября 2016 г. |
|
|
20-23 сентября 2016 г. |
|
|
20-23 сентября 2016 г. |
|
|
26-30 сентября 2016 г. |
|
|
September 26-30, 2016 |
|
|
27-28 сентября 2016 г. |
|
|
October 2-6, 2016 |
http://conf.nsc.ru/MCR-X/en ; |
|
October 2-7, 2016 |
|
|
3-7 октября 2016 г. |
|
|
3-7 октября 2016 г. |
mkiskin@igic.ras.ru |
|
October 3-7, 2016 |
|
|
4-5 октября 2016 г. |
|
|
4-7 октября 2016 г. |
|
|
9-14 октября 2016 г. |
|
|
October 11-13, 2016 |
|
|
October 12-14, 2016 |
|
|
18-21 октября 2016 г. |
|
|
18-21 октября 2016 г. |
|
|
October 23-26, 2016 |
|
|
23-27 октября 2016 г. |
|
|
30 октября – 2 ноября 2016 г. |
|
|
1-3 ноября 2016 г. |
|
|
3-5 ноября 2016 г. |
|
|
November 11-13, 2016 |
|
|
14-16 ноября 2016 г. |
sshavshukova@mail.ru |
|
14-18 ноября 2016 г. |
|
|
17-18 ноября 2016 г. |
http://plastpolymer.su |
|
21-26 ноября 2016 г. |
|
|
22-25 ноября 2016 г. |
|
|
November 27 – December 2, 2016 |
|
|
28-30 ноября 2016 г. |
|
|
|
|
|
January 17-21, 2017 |
|
|
March 2-3, 2017 |
|
|
22-26 мая 2017 г. |
|
|
5-10 июня 2017 г. |
|
|
August 27 – September 1, 2017 |
|

